Description
Finds the atoms within a sphere with the indicated center and radius
Usage
ball(pdb, res, chain, r, backbone = FALSE)
Arguments
pdb is either a PDB id, or the path to a pdb file.
chain a character indicating the chain to which the residue belongs
res position in the primary structure of the residue of interest, which will be use as center of the sphere.
r radious in ångströms of the sphere.
backbone logical, when TRUE it means that we include those atoms belonging to the main chain (CA, N, O and C) beside all the side chain atoms.
Value
A dataframe with the atoms identification and their distances to the central atom.
See Also
pairwise.dist(), res.dist(), dist2closest()
Details
The continuous formation and rupture of non-covalent bonds (intra- and intermolecular), gives rise to dynamic molecular interactions, which are cosubstantial to life. These non-covalent interactions can adopt many forms, but all of them are fundamentally electrostatic in nature, and strongly influenced by distances.
| Type of interaction | Energy dependence on the distance |
|---|---|
| Ion-ion |  |
| Ion-dipole | 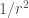 |
| Dipole-dipole |  |
| Ion-induced dipole |  |
| Dipole-indiced dipole | 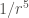 |
| Dispersion | 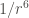 |
Therefore, computing spatial distances (in ångströms, Å), either between atoms or residues, is a useful task in many different contexts. A number of function from the package ptm will help us in these tasks:
- res.dist
- dist2closest
- ball (the current document)
- pairwise.dist
There is a set of atoms of the protein molecule of particular interst because of they intramolecular interactions with other atoms that may be close enough. The types of relevant atoms that can be study with the function ball() are:
- Sulfur delta (SD) from Met residues.
- Sulfur gamma (SG) from Cys residues.
- Oxigen gamma (OG) from Ser residues.
- Oxigen gamma-1 (OG1) from Thr residues.
- Carboxylate atoms (OE1 and OE2) from Glu residues.
- Carboxylate atoms (OD1 and OD2 ) from Asp residues.
- Nitrogen atoms (ND1 and NE2) of the imidazol from His residues.
- Nitrogen atoms (NE, NH1 and NH2) of the guanidinum from Arg residues.
- Nitrogen zeta (NZ) from Lys residues.
- Benzene ring atoms (CG, CD1, CD2, CE1, CE2 and CZ) from Phe residues.
- Benzene ring atoms (CG, CD1, CD2, CE1, CE2 and CZ) from Tyr residues.
- Indol ring atoms (ring-1: CG, CD1, CD2, NE1, CE2; ring-2: CD2, CE3, CZ2, CH2, CZ3, CE2) from Trp residues.
The function ball() takes the (x, y, z) coordinates of the chosen atom (or centroid of the functional group) as the center of a sphere (ball) of radious \(r\), and finds all the atoms from the protein that are within this sphere. For instance, if we are interested in the interactions of the SD from Met181 of the protein human inositol polyphosphate multikinase (PDB ID: 6R7F), then we may procedure:
kable(ball(pdb = '6e7f', chain = 'A', res = 181, r = 4.5))
| elety | resid | chain | resno | x | y | z | distance | |
|---|---|---|---|---|---|---|---|---|
| 638 | SD | MET | A | 143 | -18.059 | 7.818 | 23.615 | 4.338562 |
| 953 | CG2 | VAL | A | 183 | -13.143 | 3.458 | 20.494 | 4.433208 |
| 1046 | CG | GLU | A | 194 | -13.272 | 1.839 | 26.725 | 4.387182 |
| 1086 | CE2 | TYR | A | 198 | -13.862 | 5.293 | 28.560 | 4.358599 |
| 1196 | CB | ARG | A | 213 | -10.772 | 6.803 | 25.020 | 4.269295 |
| 1197 | CG | ARG | A | 213 | -10.420 | 5.329 | 24.811 | 4.275447 |
| 1207 | CB | PHE | A | 214 | -14.851 | 7.520 | 21.619 | 3.529871 |
| 1208 | CG | PHE | A | 214 | -15.772 | 7.276 | 20.445 | 4.495141 |
It should be noted that all atoms belonging to the central residue, in this case Met181, are excluded in the returned dataframe. Also, keep in mind that if we select a central residue that doesn’t belong to the set {Met, Cys, Ser, Thr, Glu, Asp, His, Arg, Lys, Phe, Tyr, Trp}, the function will return an error message.
