Description
Represents the sensitivity of a given position to changes in stability of a protein (ΔΔG).
Usage
ddG.profile(prot, ch, pos, pH = 7, Te = 25)
Arguments
prot either the 4-letter identifier of a PDB structure, or the amino acid sequence (one letter amino acid code) of a protein.
ch a letter identifying the chain of interest.
pos the position, in the primary structure, of the residue to be mutated.
pH a numeric value between 0 and 14.
Te a numeric value indicating the temperature in degrees Celsius.
Value
The function returns a dataframe with the ΔΔG values (kcal/mol) for each alternative amino acid, and a barplot grouping the amino acids according to their physicochemical nature.
Details
Thermodynamic stability is a fundamental property of proteins that influences protein structure, function, expression, and solubility. Not surprisingly, prediction of the effect of point mutations on protein stability is a topic of great interest in many areas of biological research, including the field of protein post-translational modifications. Consequently, a wide range of strategies for estimating protein energetics has been developed. In the package ptm we have implemented two popular computational approaches for prediction of the effect of amino acid changes on protein stability.
On the one hand, foldx.mut() implements FoldX (buildmodel and positionscan methods), a computational approach that uses a force field method and athough it has been proved to be satisfactorily accurate, it is also a time-consuming method. On the other hand, I-Mutant is a method based on machine-learning and it represents an alternative much faster to FoldX. The ptm function that implements this last approach is imutant().
Although using very different strategies, both functions assess
the thermodynamic stability effect of substituting a single amino acid residue in the protein of interest. To do that, both function perform an estimation of the change in Gibbs free energy, 

Where 
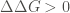

To illustrate the use of ddG.profile(), a function that offers the possibility to plot the stability profile at a given position within the primary structure of a protein of interest, we will use as a model the β1 domain of streptococcal protein G (Gβ1), which has been extensively characterized experimentally in terms of thermodynamic stability. More concretely, we are going to address the effect of changing residues at the positions 26 and 27 of the β1 domain of streptococcal protein G, where we find alanine (A) and glutamate (E), respectively in the wild type form of the Gβ1 protein.
E27 <- ddG.profile('1pga', 'A', pos = 27)
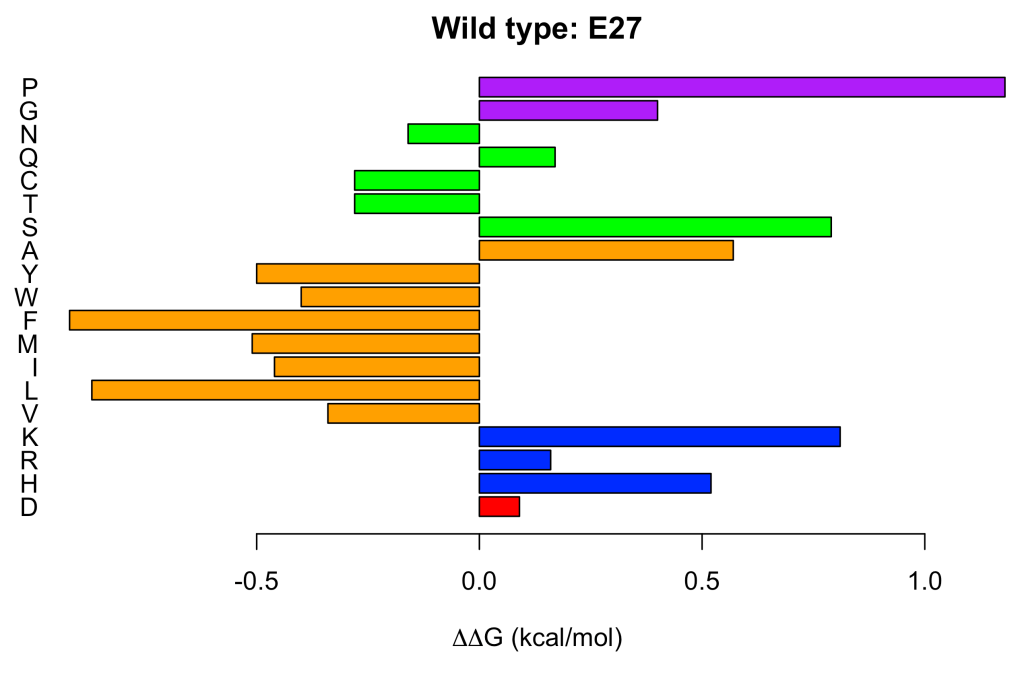
Each amino acid has been colored according to its physicochemical nature. Acidic (E, D) in red, basic (H, R, K) in blue, hydrophobic (L, I, M, F, W, Y, A) in orange, polar (S, T, C, Q, N) in green and special (G, P) in purple.
It can be seen at a glance, that hydrophobic amino acids, with the excepticon of alanine (A), tend to increase the protein stability when replacing to glutamate (E) at position 27. This results might seem surprising, but it is in perfect agreement with the experimental observations (Nisthal et al. 2019). Less surprising and equally in line with the experimental evidence, is that proline (P) is the worst (most destabilizing) amino acid for incorporation at position 27.
When we address the stability profile for position 26, the result is quite different:
A26 <- ddG.profile('1pga', 'A', pos = 26)

We conclude that any substitution at this position is destabilizing, regardless the nature of the amino acid being introduced. This conclusion is again in excelent agreement with the experimental results (Nisthal et al. 2019).
