Description
Computes Folding Free Energy (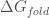
Usage
foldx.stab(pdb, pH = 7, I = 0.05)
Arguments
pdb the 4-letter identifier of a PDB structure or the path to a PDB file.
pH a numeric value between 0 and 14.
I a value indicating the molar ionic strength.
Value
The function computes and returns the ΔG (kcal/mol) of the folding process of the requested protein.
References
Schymkowitz et al (2005) Nucl. Ac. Res. 33:W382-W388.
Details
Thermodynamic stability is a fundamental property of proteins that influences protein structure, function, expression, and solubility. Not surprinsinly, prediction of the effect of point mutations on protein stability is a topic of great interest in many areas of biological research, including the field of protein post-translational modifications. Consequentely, a wide range of strategies for estimating protein energetics has been developed. In the package ptm we have implemented several commands of the FoldX suite. One of these FoldX’s commands is Stability that is implemented in the function foldx.stab(). This function computes the ΔG, in kcal/mol, of the folding process.
Therefore, the more negative this values is, the more stable the protein is. Nevertheless, we must be cautions when using foldx.stab() because
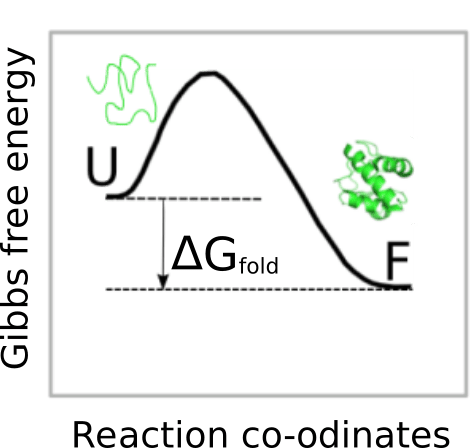
Since the functions based on FoldX commands show the highest accuracy when the energy difference can be calculated, foldx.stab() may be useful to compare the thermodynamic stability of two conformations of the same protein. For instance, Calmodulin (CaM) is a multifunctional calcium-binding protein expressed in all eukaryotic cells. The binding of the secondary messenger Ca2+ to CaM changes the CaM conformation and its affinity for its various targets. In a Ca2+ free medium, CaM exhibits a spatial conformation corresponding to the so-called apoCaM (PDB ID: 1CFD). On the other hand, when CaM binds two Ca2+ ions, the protein adopts a different spatial conformation corresponding to the holoCaM (PDB ID: 1CLL). We can compute the change in Gibbs free energy for the conformational change from apoCaM to holoCaM using foldx.stab() as indicated in the next figure.
To perform this calculations we can run the following chunk:
apo <- foldx.stab('1cfd')
holo <- foldx.stab('1cll')
G_conf <- holo - apo
G_conf
## [1] -46.2534 ## attr(,"pdb") ## [1] "1cll" ## attr(,"units") ## [1] "kcal/mol"
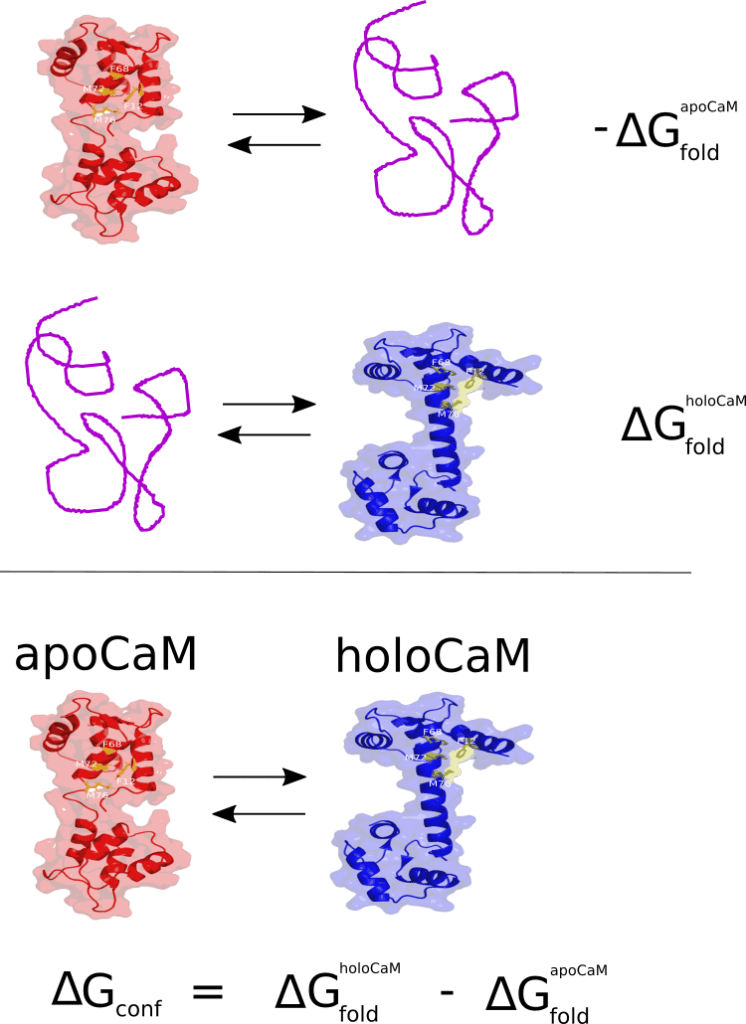
The function foldx.stab() also allows us to illustrate the effect of the ionic strength on the stability of proteins. To this end we will use bacteriophage T4 lysozyme (7LZM). Electrostatic interactions, also known as salt bridges or ionic interactions, are formed between groups found in basic (Arg, Lys and His) and acidic (Glu, Asp) residues of proteins at neutral pH. In the figure below the basic (blue) and acidic (red) amino acids find in lysozyme are show as a mesh.
The strength of these interactions is given by the Coulomb’s law:

where 
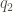


Next we compute and plot the thermodynamic stability of lysozyme at high, medium and low ionic strengths:
high <- foldx.stab("7lzm", I = 1)
medium <- foldx.stab("7lzm", I = 0.05)
low <- foldx.stab("7lzm", I = 0.0001)
I <- c(1, 0.05, 0.0001)
DG <- c(high, medium, low)
plot(-log(I),DG, ty = "b", ylab = expression(paste(Delta, 'G ', "(kcal/mol)")))
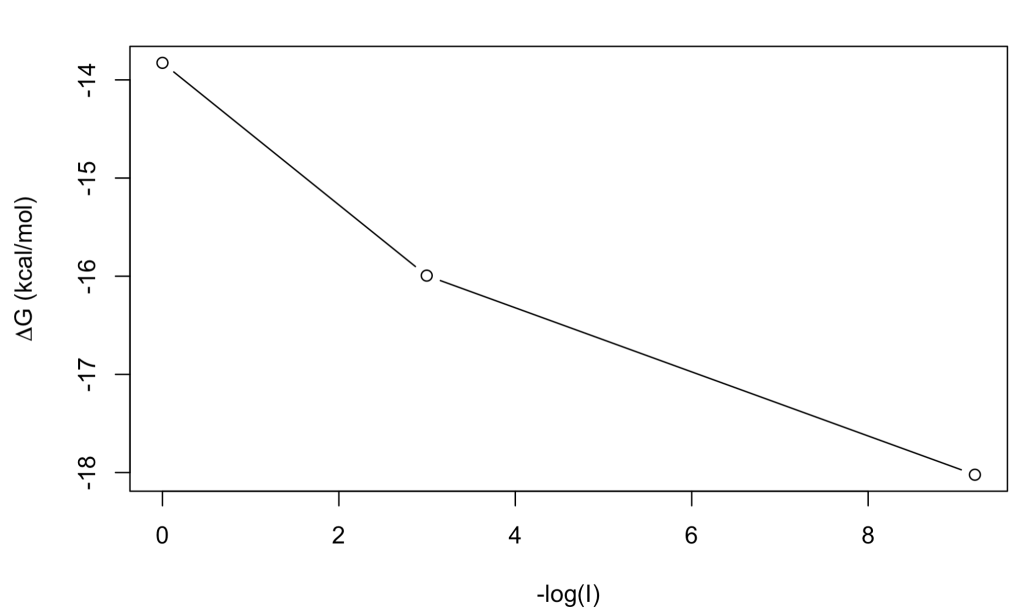
At low ionic strength the stability is higher (ΔG = -18 kcal/mol) than at high ionic strength (ΔG = -13.8 kcal/mol), which may be in line with stronger salt bridges, although this sort of simple interpretations are a risky business when dealing with protein stability!
