Description
Explores the relationship among proteins from a given set
Usage
net.go(data, threshold = 0.2, silent = FALSE)
Arguments
data either a vector containing the UniProt IDs (vertices) or the path to the txt or rda file containing them.
threshold threshold value of the Jaccard index above which two proteins are considered to be linked.
silent logical, if FALSE print details of the running process.
Value
This function first searches the GO for each vertex and then computes the Jaccard index for each protein pair based on their GO terms. Afterwards, an adjacency matrix is computed, where two proteins are linked if their Jaccard index is greater than the selected threshold. The function returns a list containing (i) the dataframe corresponding to the computed Jaccard matrix, (ii) the adjacency matrix, (iii) a vector containing the vertices, and (iv) a matrix describing the edges of the network.
References
Aledo & Aledo (2020) Antioxidants 9(10), 987.
Rhee et al. (2008) Nature Reviews Genetics 9:509–515.
See Also
search.go(), term.go(), get.go(), background.go(), gorilla(), hdfisher.go()
Details
The Gene Ontolory project (GO) provides a controlled vocabulary to describe gene and gene product attributes. Thus, a GO annotation is a statement describing some biological aspect. GO annotations are created by associating a gene or gene product with a GO term. Together, these statements comprise a “snapshot” of current biological knowledge. Hence, GO annotations capture statements about three disjoint categories: cellular component, molecular function and biological process. In this way, GO terms, using a controlled and hierarchical vocabulary, help to describe how a gene functions at the molecular level, where in the cell it functions, and what biological processes it helps to carry out.
The use of this vocabulary (GO annotations) has diverse applications, perhaps the most popular of them is the functional profiling. The goal of functional profiling is to determine which processes might be different in particular sets of genes (or gene products). That is, GO annotations are used to determine which biological processes, functions, and/or locations are significantly over- or under-represented in a given group of genes (or gene products). This approach is also known as GO term enrichment analysis.
Another popular application of GO annotations is GO network analysis, that we address herein. To formalize the analysis, we began by defining the sets P and O in the following way.



That is, P is going to be the set of all the proteins we want to analyze. Each element from P is identified by its corresponding UniProt ID. For instance, let’s P be the set of all human proteins containing methionine sulfoxide (MetO) after an oxidative challenge to the cell. On the other hand, O is formed by all the GO terms of human proteins. In this way, we can define the function f as follows:
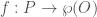

where 


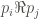

Formally, a graph 
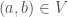
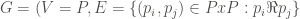
Thus, our first goal is to get the set we have called P:
sites <- meto.search(organism = 'Homo sapiens',
oxidant = 'hydrogen peroxide')
P <- unique(sites$prot_id[which(sites$met_vivo_vitro == "vivo")])
Once we have the set P, we are ready to use the function net.go():
network <- net.go(P, threshold = 0.5, silent = TRUE)
This function first searchs the GO terms for each node (protein in P) and then computes the Jaccard index for each protein pair based on their GO terms. Afterwards, an adjacency matrix is computed, where two proteins are linked if their Jaccard index is greater than the selected threshold.
Let’s examine the first 10 columns and rows from the Jaccard matrix we have obtained:
Jaccard <- network[[1]]
Jaccard[1:10, 1:10]
## A2RRP1 A3KN83 A5YKK6 A6NEC2 A6NIZ1 A8K0Z3 A8MWD9 B0I1T2 O00116 O00161 ## A2RRP1 NA 0 0.071 0.045 0.150 0.100 0.000 0.043 0.071 0.075 ## A3KN83 NA NA 0.025 0.000 0.000 0.000 0.040 0.000 0.000 0.000 ## A5YKK6 NA NA NA 0.022 0.070 0.063 0.039 0.029 0.082 0.048 ## A6NEC2 NA NA NA NA 0.043 0.023 0.000 0.043 0.000 0.048 ## A6NIZ1 NA NA NA NA NA 0.071 0.000 0.089 0.069 0.073 ## A8K0Z3 NA NA NA NA NA NA 0.000 0.061 0.061 0.083 ## A8MWD9 NA NA NA NA NA NA NA 0.000 0.000 0.000 ## B0I1T2 NA NA NA NA NA NA NA NA 0.018 0.078 ## O00116 NA NA NA NA NA NA NA NA NA 0.062 ## O00161 NA NA NA NA NA NA NA NA NA NA
As expected, we note that Jaccard is an upper triangular matrix. Now we also examine the first rows and columns from the adjacency matrix:
Adjacency <- network[[2]]
Adjacency[1:10, 1:10]
## A2RRP1 A3KN83 A5YKK6 A6NEC2 A6NIZ1 A8K0Z3 A8MWD9 B0I1T2 O00116 O00161 ## A2RRP1 0 0 0 0 0 0 0 0 0 0 ## A3KN83 0 0 0 0 0 0 0 0 0 0 ## A5YKK6 0 0 0 0 0 0 0 0 0 0 ## A6NEC2 0 0 0 0 0 0 0 0 0 0 ## A6NIZ1 0 0 0 0 0 0 0 0 0 0 ## A8K0Z3 0 0 0 0 0 0 0 0 0 0 ## A8MWD9 0 0 0 0 0 0 0 0 0 0 ## B0I1T2 0 0 0 0 0 0 0 0 0 0 ## O00116 0 0 0 0 0 0 0 0 0 0 ## O00161 0 0 0 0 0 0 0 0 0 0
Not surprisingly, this submatrix is the zero matrix. That is, any of the 10 first proteins are related among them according to our criteria (threshold = 0.5). Nevertheless, we can check that our graph will have 578 edges (pairs of related proteins):
edges <- network[[4]]
nrow(edges)
## [1] 578
head(edges)
## [,1] [,2] ## [1,] "A3KN83" "O15037" ## [2,] "A5YKK6" "Q9NZN8" ## [3,] "A6NEC2" "O75976" ## [4,] "A6NEC2" "Q5JRX3" ## [5,] "A6NEC2" "Q9NZ08" ## [6,] "A6NIZ1" "P84085"
Now, if we have previously installed the package igraph, then we are in conditions to plot our network:
G <- igraph::graph_from_data_frame(d = edges,
vertices = network[[3]],
directed = FALSE)
plot(G,
vertex.label = NA,
edge.color = "black",
vertex.size = 0.5 + (igraph::degree(G))^(1/5),
layout = igraph::layout_components(G)
)

Other functions related to GO offered by ptm are:
search.go
term.go
get.go
bg.go
hdfisher.go
gorilla
net.go (current doc)
