Description
Parses a HSSP file to return dataframes
Usage
parse.hssp(file, keepfiles = TRUE)
Arguments
file input hssp file.
keepfiles logical, if TRUE the dataframes will be saved in the working directory and we will keep the hssp file.
Value
Returns 4 dataframes containing the information found in hssp files, as describe below.
References
Touw et al (2015) Nucl. Ac. Res. 43:D364-368.
Lange et al (2020) Protein Sci. 29:330-344.
See Also
msa(), custom.aln(), list.hom(), get.hssp(), shannon(), site.type()
Details
Multiple sequence alignment (MSA), which consists in the alignment of three or more biological sequences. From the output, homology can be inferred and the evolutionary relationships between the sequences studied. Thus, alignment is the most important stage in most evolutionary analyses. In addition, MSA is also an essential tool for protein structure and function prediction. The package ptm offers several functions that will assist you in the process of sequence analysis:
msa
custom.aln
list.hom
parse.hssp (current doc)
get.hssp
shannon
site.type
The function parse.hssp() is a parser of HSSP files. HSSP, which stands for Homology-derived Secondary Structure of Proteins, are files containing information related to MSAs of UniProtKB against PDB. When the argument ‘keepfiles’ is set to TRUE, the parse.hssp() function will build and save (in the working directory) the following 4 dataframes:
-
id_seq_list.Rda: This block of information holds the metadata per sequence, and some alignment statistics. For a detailed description of the information that can be find in this block, check here.
-
id_aln.Rda: This dataframe contains the alignment itself (each sequence is a column). Additional information such as secondary structure, SASA (solvent accessible surface area), etc is also found in this block.
-
id_profile.Rda: This dataframe holds per amino acid type its percentage in the list of residues observed at the indicated position. In addition, this dataframe also informs about the entropy at each position, as well as the number of sequences spanning this position (NOOC).
-
id_insertions.Rda: A dataframe with information regarding those sequences that contain insertions. Click here for further details.
Since, parse.hssp() is a parser, you must have in your machine the corresponding hssp file that you may have downloaded previously, for instance, using the server xssp. Thus, to illustrate herein the use of parse.hssp(), I got the file ‘3cwm.hssp’ in my current directory
profile <- parse.hssp(file = "./3cwm.hssp", keepfiles = TRUE)
The object ‘profile’ is a dataframe with as many rows as residues has the protein. For each position, the following variables (columns) are shown:
- SeqNo: Sequence residue number.
- PDBNo: PDB residue number.
- V: Percentage at which the amino acid valine (Val) is found at that position.
- L: Percentage at which the amino acid leucine (Leu) is found at that position.
- I: Percentage at which the amino acid Isoleucine (Ile) is found at that position.
- M: Percentage at which the amino acid methionine (Met) is found at that position.
- F: Percentage at which the amino acid phenylalanine (Phe) is found at that position.
- W: Percentage at which the amino acid tryptopha (Trp) is found at that position.
- Y: Percentage at which the amino acid tyrosine (Tyr) is found at that position.
- A: Percentage at which the amino acid alanine (Ala) is found at that position.
- G: Percentage at which the amino acid glycine (Gly) is found at that position.
- P: Percentage at which the amino acid proline (Pro) is found at that position.
- S: Percentage at which the amino acid serine (Ser) is found at that position.
- T: Percentage at which the amino acid threonine (Thr) is found at that position.
- C: Percentage at which the amino acid cysteine (Cys) is found at that position.
- Q: Percentage at which the amino acid glutamine (Gln) is found at that position.
- N: Percentage at which the amino acid asparragine (Asn) is found at that position.
- H: Percentage at which the amino acid histidine (His) is found at that position.
- R: Percentage at which the amino acid arginine (Arg) is found at that position.
- K: Percentage at which the amino acid lysine (Lys) is found at that position.
- E: Percentage at which the amino acid glutamate (Glu) is found at that position.
- D: Percentage at which the amino acid aspartate (Asp) is found at that position.
- NOCC: Number of aligned sequences spanning this position (including the test sequence).
- NDEL: Number of sequences with a deletion in the test protein at this position.
- NINS: Number of sequences with an insertion in the test protein at this position.
- ENTROPY: Entropy measure of sequence variaparbility at this position.
- RELENT: Relative entropy, i.e. entropy normalized to the range 0-100.
- WEIGHT: Conservation weight.
We can have a visual impression of which are the most variable and the most conserved positions by plotting the relative entropy as a function of the position:
plot(profile$SeqNo, profile$RELENT, ty = 'h', xlab = 'Position', ylab = 'Relative Entropy')
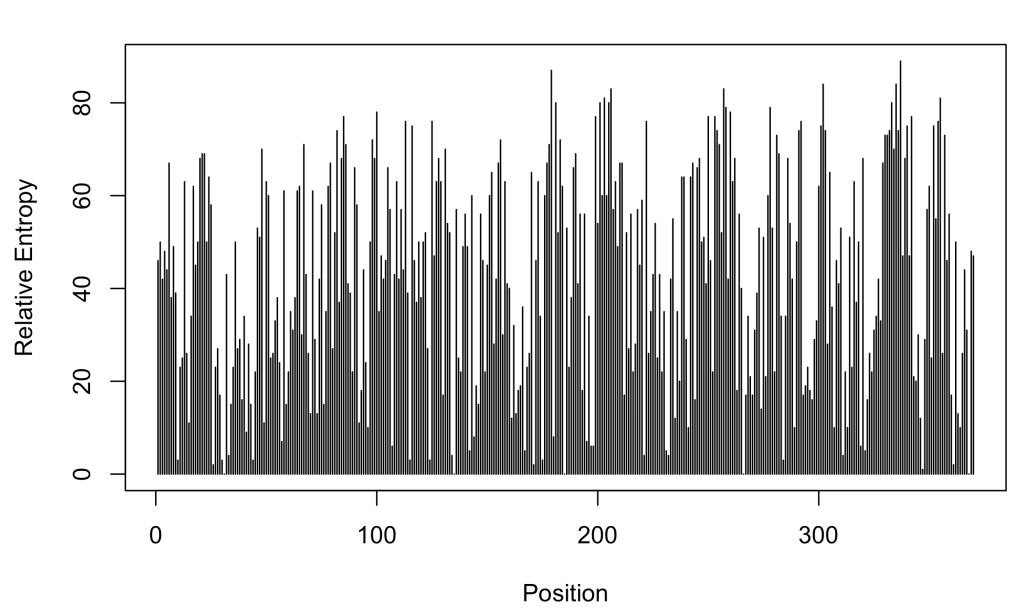
In this way, the most variable position is:
maxS_at <- which(profile$ENTROPY == max(profile$ENTROPY))
x <- as.data.frame(t(profile[maxS_at, 3:22]))
x$col <- c(rep("orange", 8), rep("purple", 2), rep("green", 5), rep("blue", 3), rep("red",2))
names(x) <- c('frequency', 'col')
barplot(height = x$frequency,
names = rownames(x),
col = x$col,
main = paste("Position:", profile$PDBNo[maxS_at]))

Here, we have colored the amino acids according to their physicochemical nature. Acidic (E, D) in red, basic (H, R, K) in blue, hydrophobic (L, I, M, F, W, Y, A) in orange, polar (S, T, C, Q, N) in green and special (G, P) in purple. We observe that, except Trp and Cys, any amino acid can be found at this position.
In contrast, the most conserved position is:
minS_at <- which(profile$ENTROPY == min(profile$ENTROPY))
x <- as.data.frame(t(profile[minS_at, 3:22]))
x$col <- c(rep("orange", 8), rep("purple", 2), rep("green", 5), rep("blue", 3), rep("red",2))
names(x) <- c('frequency', 'col')
barplot(height = x$frequency,
names = rownames(x),
col = x$col,
main = paste("Position:", profile$PDBNo[minS_at]))
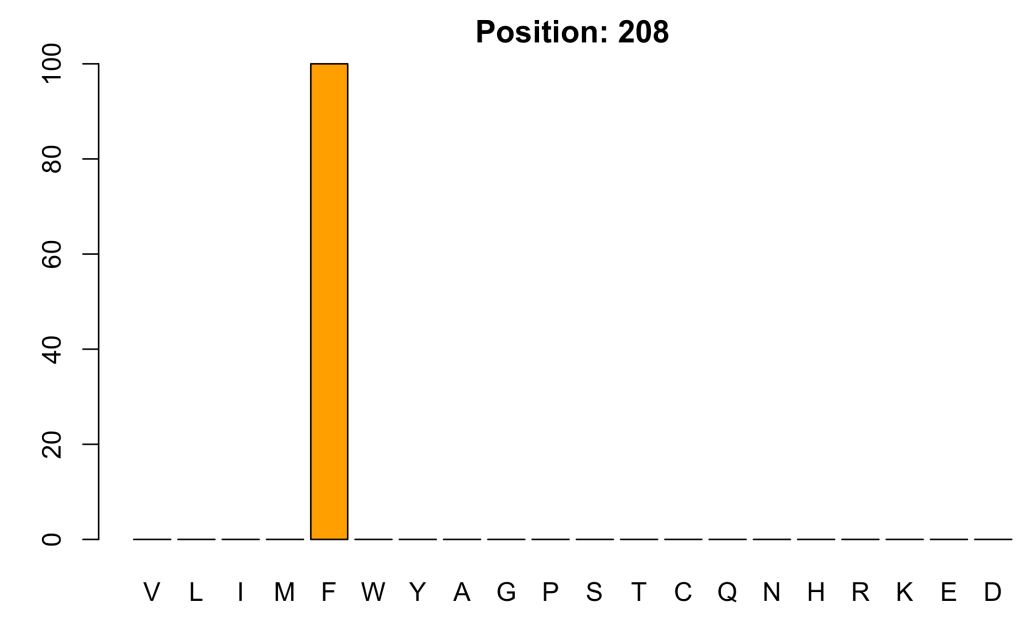
where phenylalanine is the only amino acid present!
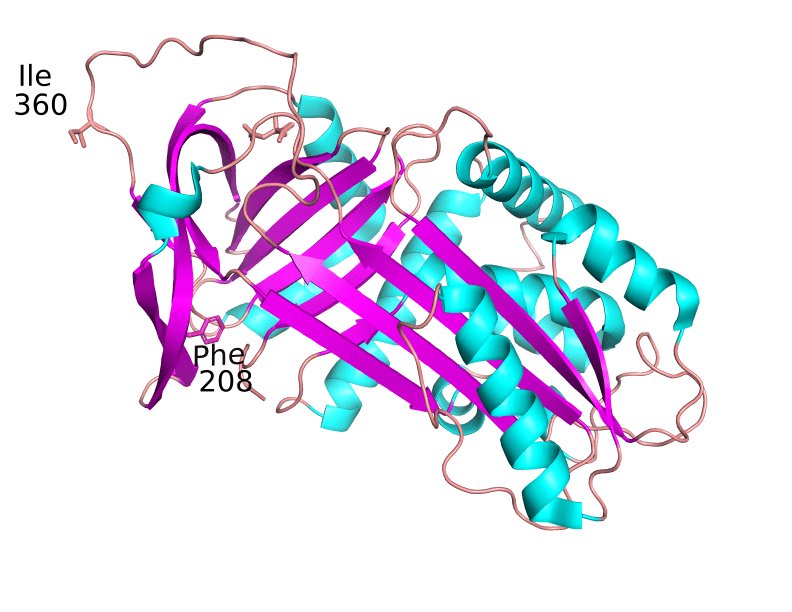
The 3D structure of the human protein is shown below. The conserved Phe208 and the highly variable position 360, that in the reference protein (PDB ID: 3CWM) is occupied by Ile, are marked.
In addition to this dataframe we have called ‘profile’, we can access, if we wish, the alignment itself:
load("./3cwm_aln.Rda")
dim(aln)
[1] 370 1205
This dataframe, that we have placed in an object named ‘aln’, has 370 raws (one per residue) and 1205 columns. The first eight columns are:
- SeqNo: Sequence residue number.
- PDBNo: PDB residue number.
- Chain: Chain identifier.
- AA: Amino Acid at that position in the reference sequence.
- SS: Element of secondary structure.
- ACC: Solvent accessible area.
- NOCC: Number of aligned sequences spanning this position (including the reference sequence).
- VAR: Sequence variability on a scale of 0-100 as derived from the number of sequences aligned.
The ninth column (named in this example ‘P01009’) gives the reference sequence, while the remaining columns provide the sequence of the protein included in the alignment. These columns are named with the UniProt ID of the corresponding protein.
Information regarding the metadata per sequence, and some alignment statistics, can be found in a third dataframe:
load("./3cwm_seq_list.Rda")
head(seq_list)
## NR ID IDE WSIM IFIR ILAS JFIR JLAS LALI NGAP LGAP LESEQ2 ACCNUM ## 1 1 A1AT_HUMAN 1.00 1.00 1 370 48 417 370 0 0 418 P01009 ## 2 2 E9KL23_HUMAN 1.00 1.00 1 370 48 417 370 0 0 418 E9KL23 ## 3 3 A0A024R6I7_H 0.99 0.99 1 370 48 417 370 0 0 418 A0A024R6I7 ## 4 4 A0A0G2JRN3_H 0.99 0.99 1 308 48 355 308 0 0 359 A0A0G2JRN3 ## 5 5 A0A2J8QMJ1_P 0.99 0.99 1 370 48 417 370 0 0 418 A0A2J8QMJ1 ## 6 6 A0A2J8QMJ5_P 0.99 0.99 1 308 48 355 308 0 0 359 A0A2J8QMJ5 ## PROTEIN ## 1 Alpha-1-antitrypsin OS=Homo sapiens OX=9606 GN=SERPINA1 PE=1 SV=3 ## 2 Epididymis secretory sperm binding protein Li 44a OS=Homo sapiens OX=9606 GN=SERPINA1 PE=2 SV=1 ## 3 Alpha-1-antitrypsin OS=Homo sapiens OX=9606 GN=SERPINA1 PE=1 SV=1 ## 4 Alpha-1-antitrypsin OS=Homo sapiens OX=9606 GN=SERPINA1 PE=1 SV=1 ## 5 SERPINA1 isoform 1 OS=Pan troglodytes OX=9598 GN=SERPINA1 PE=3 SV=1 ## 6 SERPINA1 isoform 19 OS=Pan troglodytes OX=9598 GN=CK820_G0025082 PE=3 SV=1
The number of rows, in our example, is 1197 (one per sequence included in the alignment). The variables (columns) holded in this dataframe are:
- NR: Sequence number.
- ID: EMBL/SWISSPROT identifier of the aligned (homologous) protein.
- IDE: Percentage of residue identity of the alignment.
- WSIM: Weighted similarity of the alignment.
- IFIR: First residue of the alignment in the test sequence.
- ILAS: Last residue of the alignment in the test sequence.
- JFIR: First residue of the alignment in the alignend protein.
- JLAS: Last residue of the alignment in the alignend protein.
- LALI: Length of the alignment excluding insertions and deletions.
- NGAP: Number of insertions and deletions in the alignment.
- LGAP: Total length of all insertions and deletions.
- LSEQ2: Length of the entire sequence of the aligned protein.
- ACCNUM: SwissProt accession number.
- PROTEIN: One-line description of aligned protein.
Finally, a fourth dataframe, named ‘insertions’ can be assessed
load("./3cwm_insertions.Rda")
inser
## AliNo IPOS JPOS Len Sequence
## 1 69 252 302 1 nTs
## 2 85 355 397 1 rQt
## 3 97 355 397 1 rQt
## 4 121 298 348 1 gVt
## 5 163 169 219 53 kVSIATAFAMPSLGAKGDARTEIMKALGYNSKKALNADVHGGVHHLLDISIRQDg
## 6 164 298 362 1 gIt
## 7 165 298 339 1 gIt
## 8 166 298 339 1 gIt
## 9 168 298 339 1 gIt
## 10 169 298 339 1 gIt
## 11 170 298 339 1 gIt
## 12 171 298 338 1 gIt
## 13 174 298 339 1 gIt
## 14 176 298 339 1 gIt
## 15 177 298 339 1 gIt
## 16 178 298 339 1 gIt
## 17 198 298 339 1 gIt
## 18 207 336 387 2 mHCw
## 19 213 336 339 3 gASMd
## 20 217 211 277 2 rDNe
## 21 222 336 477 5 gSCFSYv
## 22 230 336 348 4 lSAPFp
## 23 233 298 333 1 gVt
## 24 234 336 393 3 gASMd
## 25 250 336 448 6 xXXALLYv
## 26 250 339 457 8 gSCSYAAPPp
## 27 253 252 287 1 nRr
## 28 256 336 391 4 mSAPId
## 29 258 336 392 4 fSAEFp
## 30 260 336 348 4 lSAQFp
## 31 262 336 348 4 lSAGFp
## 32 268 336 336 4 rSGDFp
## 33 270 336 348 4 mSAEIp
## 34 271 336 348 4 lSAEFp
## 35 273 336 348 4 ySIEIp
## 36 276 336 348 4 lSAEIp
## 37 278 336 346 4 lSAEFp
## 38 281 336 348 4 lTAEFp
## 39 282 336 338 4 rSGDFp
## 40 284 336 336 4 rSGDFp
## 41 285 336 348 4 mSAEFp
## 42 286 336 348 4 lSALFp
## 43 288 336 348 4 lSAEFp
## 44 290 336 346 4 lSAMFp
## 45 291 336 335 4 rSGDFp
## 46 294 336 348 4 lSRRFp
## 47 297 336 348 4 lSAVIp
## 48 300 336 348 4 mSLPIp
## 49 301 336 348 4 mSAQIp
## 50 306 200 244 1 sIe
## 51 306 252 297 1 kWk
## 52 306 336 382 3 tSAKl
## 53 307 260 362 2 sEDl
## 54 309 336 392 4 rSGDFp
## 55 317 308 369 2 sKVr
## 56 322 24 70 2 dGKs
## 57 323 24 90 2 dGEs
## 58 326 308 329 2 sKVr
## 59 328 200 203 1 tQk
## 60 328 336 340 4 kSAPFi
## 61 330 336 383 4 vCQRNr
## 62 331 336 387 3 fSAMv
## 63 334 336 392 4 rSGQIs
## 64 336 226 277 2 rNVa
## 65 336 252 305 1 rLe
## 66 336 260 314 1 rSr
## 67 340 336 336 4 rSGDFp
## 68 342 336 348 4 fLLPFp
## 69 343 336 348 4 mSAVIp
## 70 344 336 346 4 vSALSp
## 71 345 336 348 4 lSAAYp
## 72 347 336 336 4 kSGDIp
## 73 348 24 36 1 gNn
## 74 348 336 346 4 mSAHFh
## 75 349 336 348 4 lLASFp
## 76 351 336 336 4 rSGDFp
## 77 353 336 343 4 lSAMFp
## 78 354 336 348 4 lSALFp
## 79 355 336 348 4 lSAEFp
## 80 356 336 348 4 mSAQIp
## 81 362 336 336 4 rSGDFp
## 82 363 336 346 4 lSAAFp
## 83 365 336 348 4 lSAQFp
## 84 366 336 348 4 lSAEFp
## 85 367 336 332 4 rSGDFp
## 86 369 211 291 1 dEe
## 87 369 260 341 1 rWv
## 88 369 336 418 4 mSGKIg
## 89 372 336 386 4 mSAPFp
## 90 374 336 405 3 fSAMi
## 91 375 211 253 1 dEe
## 92 375 260 303 1 rLi
## 93 375 336 380 1 lSs
## 94 376 308 317 2 sKVr
## 95 379 169 208 13 kGKIIFILNHIPSTh
## 96 380 336 389 4 mSFSSp
## 97 381 336 392 4 rSGDFp
## 98 383 336 414 4 rSGDFp
## 99 384 336 336 4 rSGDFp
## 100 387 336 348 5 mSSMFEt
## 101 388 336 331 4 rSGDFp
## 102 389 336 327 4 rSGDFp
## 103 390 336 348 6 mSAGPASp
## 104 391 336 332 4 rSGDFp
## 105 392 336 348 4 tSLQLp
## 106 394 336 426 4 rSGDFp
## 107 395 336 384 1 aLs
## 108 396 336 404 4 rSGDFp
## 109 397 336 397 4 rSGDFs
## 110 399 211 260 1 dEe
## 111 399 260 310 1 rRi
## 112 399 336 387 1 sTl
## 113 400 336 386 6 tSAFAEFs
## 114 402 336 374 3 rSARl
## 115 403 211 257 1 dEe
## 116 403 260 307 1 rHi
## 117 403 336 384 4 lSGLVd
## 118 404 211 257 1 dEe
## 119 404 260 307 1 rWi
## 120 404 336 384 4 lSAKVg
## 121 405 211 257 1 dEe
## 122 405 260 307 1 rHi
## 123 405 336 384 4 lSALVd
## 124 406 336 382 3 kSAPm
## 125 407 336 382 3 kSAPm
## 126 408 127 168 1 gAt
## 127 408 211 253 1 dEv
## 128 408 260 303 1 rLi
## 129 408 336 380 4 tSSRNg
## 130 409 336 378 4 mSLPFp
## 131 411 211 254 1 dEe
## 132 411 260 304 1 rRi
## 133 411 336 381 1 tSi
## 134 414 211 263 1 dEe
## 135 414 260 313 1 rGi
## 136 414 336 390 2 tSIn
## 137 415 211 254 1 dEe
## 138 415 260 304 1 rGi
## 139 415 336 381 2 tSIn
## 140 416 211 304 1 dEe
## 141 416 260 354 1 rGi
## 142 416 336 431 4 tSINNh
## 143 418 336 384 1 aLs
## 144 419 336 385 3 fSAMi
## 145 420 211 255 1 dEe
## 146 420 260 305 1 rRi
## 147 420 336 382 1 sTl
## 148 421 211 248 1 dKw
## 149 421 336 374 3 lSAPk
## 150 422 169 219 2 kGKa
## 151 423 211 250 1 dEe
## 152 423 260 300 1 rRi
## 153 424 336 343 2 rSGr
## 154 425 211 257 1 dKe
## 155 425 336 383 4 lSAKMg
## 156 427 169 212 1 kEa
## 157 427 336 380 4 rSGDFp
## 158 428 211 254 1 dEe
## 159 428 260 304 1 rRi
## 160 428 336 381 1 tSi
## 161 429 336 374 3 rSARl
## 162 430 211 257 1 dEe
## 163 430 260 307 1 rEi
## 164 430 336 384 4 lSALVe
## 165 431 336 324 4 rSGDFp
## 166 432 336 336 4 rSGDFp
## 167 433 336 324 4 rSGEFp
## 168 434 336 333 4 rSGDFp
## 169 435 336 332 4 rSGDFp
## 170 437 336 348 4 lSARFp
## 171 438 336 329 4 rSGEFp
## 172 440 336 336 4 rSGDFp
## 173 441 336 374 3 rSARl
## 174 442 211 282 1 dEe
## 175 442 260 332 1 rHi
## 176 442 336 409 3 lSGFv
## 177 442 339 415 1 pKi
## 178 443 211 282 1 dEe
## 179 443 260 332 1 rHi
## 180 443 336 409 3 lSAFv
## 181 443 339 415 1 pKi
## 182 444 211 265 1 dEe
## 183 444 260 315 1 rRi
## 184 444 336 392 3 tSGKi
## 185 445 336 392 4 rSGDFp
## 186 446 336 383 1 vHq
## 187 447 211 255 1 dEe
## 188 447 260 305 1 rVi
## 189 447 336 382 3 kSRKw
## 190 448 336 383 4 lFAAFp
## 191 450 211 260 1 dEe
## 192 450 260 310 1 rRi
## 193 450 336 387 1 tSi
## 194 451 211 300 1 dEe
## 195 451 260 350 1 rRi
## 196 451 339 427 4 tSIEFl
## 197 452 336 383 3 rSARl
## 198 453 211 257 1 dEe
## 199 453 260 307 1 rEi
## 200 453 336 384 4 lSALVe
## [ reached 'max' / getOption("max.print") -- omitted 1757 rows ]
Further details regarding the information provided by this dataframe can be obtained here.
Please, mind that if the argument ‘keepfiles’ is set to FALSE, only the dataframe ‘profile’ will be returned, and the hssp file will be delated from you machine.
