Description
Computes the euclidean distance between two given residues
Usage
res.dist(file, rA, chainA, rB, chainB, backbone = F, hatoms = F)
Arguments
pdb is either a PDB id, or the path to a pdb file.
rA an integer indicating the position of the first residue.
chainA a character indicating the chain to which belong the first residue.
rB an integer indicating the position of the second residue.
chainB a character indicating the chain to which belong the second residue.
backbone logical, when TRUE it means that we include those atoms belonging to the main chain (CA, N, O and C) beside all the side chain atoms.
hatoms logical, if TRUE we include all the hydrogen atoms in the computation as long as the PDB provides their coordinates.
Value
This function returns a list of three elements, where each of these elements is, in turn, a list of three elements providing information regarding minimal, maximal and averaged distances.
See Also
pairwise.dist(), dist2closest(), ball()
Details
The continuous formation and rupture of non-covalent bonds (intra- and intermolecular), gives rise to dynamic molecular interactions, which are cosubstantial to life. These non-covalent interactions can adopt many forms, but all of them are fundamentally electrostatic in nature, and strongly influenced by distances.
| Type of interaction | Energy dependence on the distance |
|---|---|
| Ion-ion |  |
| Ion-dipole | 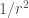 |
| Dipole-dipole |  |
| Ion-induced dipole |  |
| Dipole-indiced dipole | 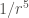 |
| Dispersion | 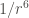 |
Therefore, computing spatial distances (in ångströms, Å), either between atoms or residues, is a useful task in many different contexts. A number of function from the package ptm will help us in these tasks:
- res.dist (current document)
- dist2closest
- ball
- pairwise.dist
The function res.dist() computes the minimal, the maximun and the average distances bewteen two given residues. Let’s take as model the alpha subunit of the human eIF2 (PDB ID 1Q8K). Ser218 and Met222
have been proposed to be functionally related. So, we’ll focus on this pair of residues.
Note that when computing the distances, we can exclude the backbone and hydrogen atoms by setting their respective parameter to FALSE.
res.dist(pdb = '1q8k',
rA = 218, chainA = 'A',
rB = 222, chainB = 'A',
backbone = FALSE, hatoms = FALSE)
## [[1]] ## [[1]][[1]] ## [1] 5.04 ## ## [[1]][[2]] ## [1] "CB-SER-218-A" ## ## [[1]][[3]] ## [1] "CB-MET-222-A" ## ## [[1]][[4]] ## [1] "minimal distance" ## ## ## [[2]] ## [[2]][[1]] ## [1] 8.37 ## ## [[2]][[2]] ## [1] "OG-SER-218-A" ## ## [[2]][[3]] ## [1] "CE-MET-222-A" ## ## [[2]][[4]] ## [1] "maximum distance" ## ## ## [[3]] ## [[3]][[1]] ## [1] 6.67 ## ## [[3]][[2]] ## [1] "SER-218-A" ## ## [[3]][[3]] ## [1] "MET-222-A" ## ## [[3]][[4]] ## [1] "average distance" ## ## ## attr(,"backbone") ## [1] FALSE
We can see that 5.04 Å is the minimal distance between these two residues. Furthermore, we see that this minimal distance is between the beta carbon from Ser218 and the beta carbon from Met222.
Similarly, we can check that the maximum distance is 8.37 Å, and corresponds to the distance between the gamma oxygen of Ser218 and the epsilon carbon of Met222.
