Description
Given a protein (PDB ID), for each methionine this function computes distances to the closest aromatic residues and the number of S-aromatic motifs formed.
Usage
saro.dist(pdb, threshold = 7, rawdata = FALSE)
Arguments
pdb either the path to the PDB file of interest or the 4-letters identifier.
threshold distance in ångströms, between the S atom and the aromatic ring centroid, used as threshold.
rawdata logical to indicate whether we also want the raw distance matrix between delta S and aromatic ring centroids.
Value
The function returns a dataframe with as many rows as methionyl residues are found in the protein. The distances in ångströms to the closest tyrosine, phenylalanine and triptophan are given in the columns, as well as the number of S-aromatic motifs detected with each of these amino acids. Also a raw distance matrix can be provided.
References
Reid, Lindley & Thornton, FEBS Lett. 1985, 190:209-213.
See Also
meto.geometry(), meto.motif()
Details
In the large inventory of the benefits provided by the presence of a sulfur atom in the side chain of methionine, we can find those effects derived from the so-called S-aromatic motifs. Indeed, an interaction of methionine and nearby aromatic residues (Phe, Tyr and Trp) was described as early as in the mid-eighties. Even earlier, a frequency of sulfur and aromatic ring in close proximity within proteins higher than expected had been noticed. Despite the potential importance of these findings, they went largely overlooked, perhaps because the physicochemical nature of this bond is only poorly understood.
Although the strength of these interactions may depend on the conditions of its environment, it is accepted that the S-aromatic interaction occurs at a greater distance (5-7 Å) than a salt bridge (< 4 Å), while the energies associated with either interaction are comparable. More recently, extensive surveys of the Protein Data Bank have revealed the importance of the methionine-aromatic motif for stabilizing protein structures and for protein-protein interactions (see the review Methionine in proteins: The Cinderella of the proteinogenic amino acids for further details).
When searching for S-aromatic motifs in proteins, two relevant variables are the distance between the delta sulfur atom (SD) and the centroid of the aromatic ring, which can be computed as the euclidean norm of the vector 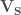
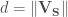
The other relevant variable is the angle theta. This angle is defined as that between the sulfur-aromatic vector (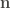

Note that two extreme cases are possible: a face-on interaction (angles around 0º) and an edge-on interaction (involving angles around 90º).
Currently, the ptm package offers three functions that may be useful for the study of these S-aromatic motifs:
- saro.dist (the current tutorial)
- saro.geometry
- saro.motif
To illustrate the use of saro.dist() we will use the alpha-1-antitrypsin protein (PDB ID: 3cwm).
a1a <- saro.dist('3cwm', threshold = 6)
## Note: Accessing on-line PDB file
kable(a1a)
| Met | Tyr_closest | Yd | number_bonds_MY | Phe_closest | Fd | number_bonds_MF | Trp_closest | Wd | number_bonds_MW |
|---|---|---|---|---|---|---|---|---|---|
| MET-A-63 | TYR-A-138 | 5.404476 | 1 | PHE-A-130 | 6.890186 | 0 | TRP-A-194 | 29.801426 | 0 |
| MET-A-220 | TYR-A-244 | 16.653779 | 0 | PHE-A-208 | 7.457474 | 0 | TRP-A-194 | 12.810557 | 0 |
| MET-A-221 | TYR-A-244 | 10.901769 | 0 | PHE-A-198 | 4.307983 | 1 | TRP-A-194 | 6.219890 | 0 |
| MET-A-226 | TYR-A-244 | 20.291255 | 0 | PHE-A-227 | 8.121467 | 0 | TRP-A-238 | 14.121028 | 0 |
| MET-A-242 | TYR-A-244 | 8.491486 | 0 | PHE-A-198 | 5.333241 | 2 | TRP-A-194 | 4.761732 | 1 |
| MET-A-351 | TYR-A-244 | 21.410995 | 0 | PHE-A-352 | 9.076933 | 0 | TRP-A-194 | 18.102479 | 0 |
| MET-A-358 | TYR-A-244 | 33.341674 | 0 | PHE-A-227 | 19.972711 | 0 | TRP-A-238 | 21.962804 | 0 |
| MET-A-374 | TYR-A-244 | 6.302081 | 0 | PHE-A-372 | 4.681723 | 2 | TRP-A-194 | 8.207798 | 0 |
| MET-A-385 | TYR-A-38 | 4.738597 | 1 | PHE-A-35 | 5.139897 | 2 | TRP-A-194 | 17.134783 | 0 |
This protein has 9 methionyl residues, thus, the a1a dataframe contains 9 rows: one per methionine residue. For each methionine residue the function returns 10 variables or features (columns of the dataframe):
- Met: the position of the methionine and the polypeptide chain identifier. For instance, Met-A-63 means the position 63 in the chain A of the protein PDB.
- Tyr_closest: the closest tyrosine to the corresponding methionine.
- Yd: distance (in ångströms) between the methionine and the closest tyrosine.
- number_bonds_MY: number of tyrosine residues at distances below the threshold.
- Phe_closest: the closest phenylalanine to the corresponding methionine.
- Fd: distance (in ångströms) between the methionine and the closest phenylalanine.
- number_bonds_MF: number of phenylalanine residues at distances below the threshold.
- Trp_closest: the closest tryptophan to the corresponding methionine.
- Wd: distance (in ångströms) between the methionine and the closest tryptophan.
- number_bonds_MW: number of tryptophan residues at distances below the threshold.
Optionally, we can get a dataframe with all the distances between methionines and aromatic residues:
met_aro <- saro.dist('3cwm', threshold = 6, rawdata = TRUE)[[2]]
## Note: Accessing on-line PDB file
met_aro <- met_aro[, -1] # removes column 1, which is not numerical
met_aro <- round(met_aro, 1) # rounds to one decimal
kable(met_aro)
| TYR-A-38 | TYR-A-138 | TYR-A-160 | TYR-A-187 | TYR-A-244 | TYR-A-297 | PHE-A-33 | PHE-A-35 | PHE-A-51 | PHE-A-52 | PHE-A-61 | PHE-A-82 | PHE-A-96 | PHE-A-119 | PHE-A-130 | PHE-A-143 | PHE-A-147 | PHE-A-182 | PHE-A-189 | PHE-A-190 | PHE-A-198 | PHE-A-208 | PHE-A-227 | PHE-A-252 | PHE-A-253 | PHE-A-275 | PHE-A-312 | PHE-A-352 | PHE-A-366 | PHE-A-370 | PHE-A-372 | PHE-A-384 | TRP-A-194 | TRP-A-238 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MET-A-63 | 25.5 | 5.4 | 11.5 | 12.1 | 25.8 | 19.6 | 15.0 | 24.4 | 20.8 | 19.0 | 9.5 | 15.3 | 10.5 | 11.7 | 6.9 | 11.9 | 16.5 | 9.8 | 21.2 | 21.6 | 36.1 | 36.4 | 39.0 | 30.2 | 30.6 | 34.0 | 14.2 | 39.1 | 35.6 | 32.0 | 25.6 | 18.2 | 29.8 | 41.1 |
| MET-A-220 | 20.5 | 38.9 | 35.7 | 30.2 | 16.7 | 24.1 | 30.5 | 25.2 | 18.0 | 23.0 | 35.7 | 37.2 | 35.3 | 38.9 | 45.1 | 41.2 | 43.7 | 42.7 | 21.5 | 19.1 | 9.0 | 7.5 | 12.6 | 11.0 | 17.5 | 20.9 | 38.4 | 20.3 | 8.5 | 8.8 | 14.0 | 20.8 | 12.8 | 13.4 |
| MET-A-221 | 21.8 | 34.6 | 30.9 | 25.5 | 10.9 | 23.5 | 29.1 | 25.3 | 15.4 | 22.1 | 33.4 | 35.0 | 31.9 | 34.8 | 41.2 | 36.2 | 40.6 | 39.6 | 15.8 | 13.4 | 4.3 | 13.2 | 11.3 | 9.8 | 18.6 | 21.6 | 37.0 | 14.9 | 11.5 | 11.4 | 13.1 | 16.9 | 6.2 | 17.9 |
| MET-A-226 | 30.6 | 43.0 | 45.2 | 39.7 | 20.3 | 37.0 | 37.6 | 29.7 | 29.0 | 32.7 | 44.4 | 40.3 | 38.8 | 49.4 | 51.7 | 50.0 | 55.6 | 52.2 | 31.7 | 25.1 | 14.6 | 25.7 | 8.1 | 18.2 | 21.2 | 18.2 | 48.9 | 11.6 | 16.8 | 22.0 | 24.5 | 28.3 | 17.1 | 14.1 |
| MET-A-242 | 20.1 | 32.6 | 32.9 | 27.4 | 8.5 | 24.6 | 26.8 | 21.0 | 16.3 | 21.1 | 32.8 | 31.2 | 28.9 | 36.9 | 40.5 | 38.0 | 42.9 | 40.1 | 19.3 | 13.0 | 5.3 | 16.2 | 5.4 | 6.4 | 13.4 | 14.4 | 37.0 | 11.3 | 8.5 | 11.3 | 12.3 | 16.1 | 4.8 | 13.3 |
| MET-A-351 | 38.8 | 42.8 | 40.9 | 36.7 | 21.4 | 40.6 | 42.6 | 38.9 | 31.5 | 37.9 | 46.6 | 45.4 | 41.4 | 46.2 | 50.7 | 45.2 | 53.4 | 51.7 | 28.0 | 24.6 | 17.9 | 32.9 | 18.8 | 25.4 | 32.5 | 31.6 | 51.6 | 9.1 | 27.4 | 29.9 | 30.1 | 29.9 | 18.1 | 29.8 |
| MET-A-358 | 41.9 | 56.7 | 58.0 | 52.5 | 33.3 | 48.8 | 50.4 | 41.8 | 41.3 | 45.1 | 57.5 | 53.5 | 52.4 | 62.2 | 65.3 | 63.0 | 68.2 | 65.3 | 43.7 | 38.1 | 25.2 | 33.8 | 20.0 | 30.2 | 32.7 | 29.7 | 61.5 | 22.7 | 26.9 | 32.3 | 36.4 | 41.3 | 29.2 | 22.0 |
| MET-A-374 | 12.2 | 22.8 | 23.5 | 17.9 | 6.3 | 14.5 | 16.2 | 13.5 | 6.3 | 10.5 | 21.6 | 21.7 | 19.1 | 26.8 | 29.9 | 28.6 | 32.4 | 29.1 | 12.8 | 6.3 | 13.9 | 16.1 | 16.3 | 6.8 | 11.1 | 15.2 | 25.9 | 20.2 | 12.6 | 10.2 | 4.7 | 5.5 | 8.2 | 18.7 |
| MET-A-385 | 4.7 | 22.0 | 26.2 | 21.2 | 15.7 | 11.7 | 10.1 | 5.1 | 10.3 | 5.7 | 17.9 | 16.5 | 16.5 | 27.7 | 28.3 | 30.6 | 31.7 | 27.0 | 20.0 | 15.1 | 21.9 | 18.2 | 22.6 | 12.7 | 9.0 | 14.2 | 21.2 | 28.5 | 15.8 | 12.9 | 8.5 | 10.8 | 17.1 | 19.6 |
