Description
Carries out a partition of the structural regions of a given protein.
Usage
stru.part(pdb, cutoff = 0.25)
Arguments
pdb is either a PDB id, or the path to a pdb file.
cutoff accessibility below which a residue is considered to be buried.
Value
A dataframe where each residue is assigned to one of the four structural groups considered.
References
Levy (2010) J. Mol. Biol. 403: 660-670.
Details
Analysis of protein commonly requires the partition of their structure into regions such as the surface, interior, or interface. Whenever you are interested into analysing such a partition in a particular protein, the following function of the ptm package may be of help:
It is always crucial to consider the structure of proteins when analyzing their biological properties. For oligomeric proteins, we can define four structural regions based on the solvent-accessible surface area (SASA) and relative accessibility (ACC) of its residues. For this purpose, the function str.partition() carries out the computation of the SASA for each amino acid in the comples (ACCc) and in the monomer (ACCm). Those residues for which DACC = (ACCm – ACCc) > 0 are designed as interface residues and can be further sorted into support, rim and core according to the following criteria:
- support: DACC > 0 & ACCm < 0.25
- rim: DACC > 0 & ACCc > 0.25
- core: ACCc < 0.25 & ACCm > 0.25
When a residue exhibits DACC = 0, this can belong to one of the following categories:
- surface: DACC = 0 & ACCc > 0.25
- interior: DACC = 0 & ACCc < 0.25
The str.partition() function uses the cutoff ACC = 0.25, but the user can change this value if desired.
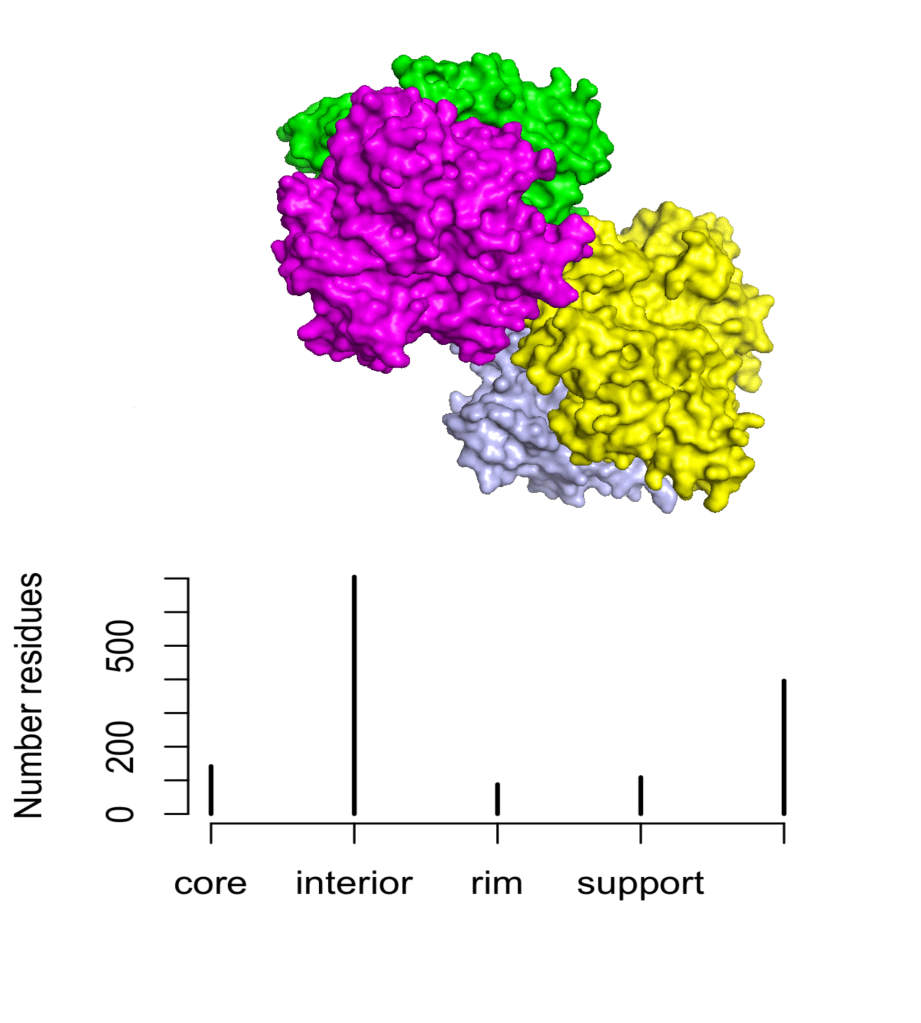
Let’s see the performance of this function with a few well known oligomeric proteins. First, we are going to deal with the proliferating cell nuclear antigen (PCNA), a homotrimeric protein.
# PCNA <- str.partition(pdb = '1AXC')
# tiff(filename = "PCNA_str", width = 10, height = 8, units = 'cm', res = 300)
# plot(table(PCNA$str), ylab = 'Number of residues')
# dev.off()
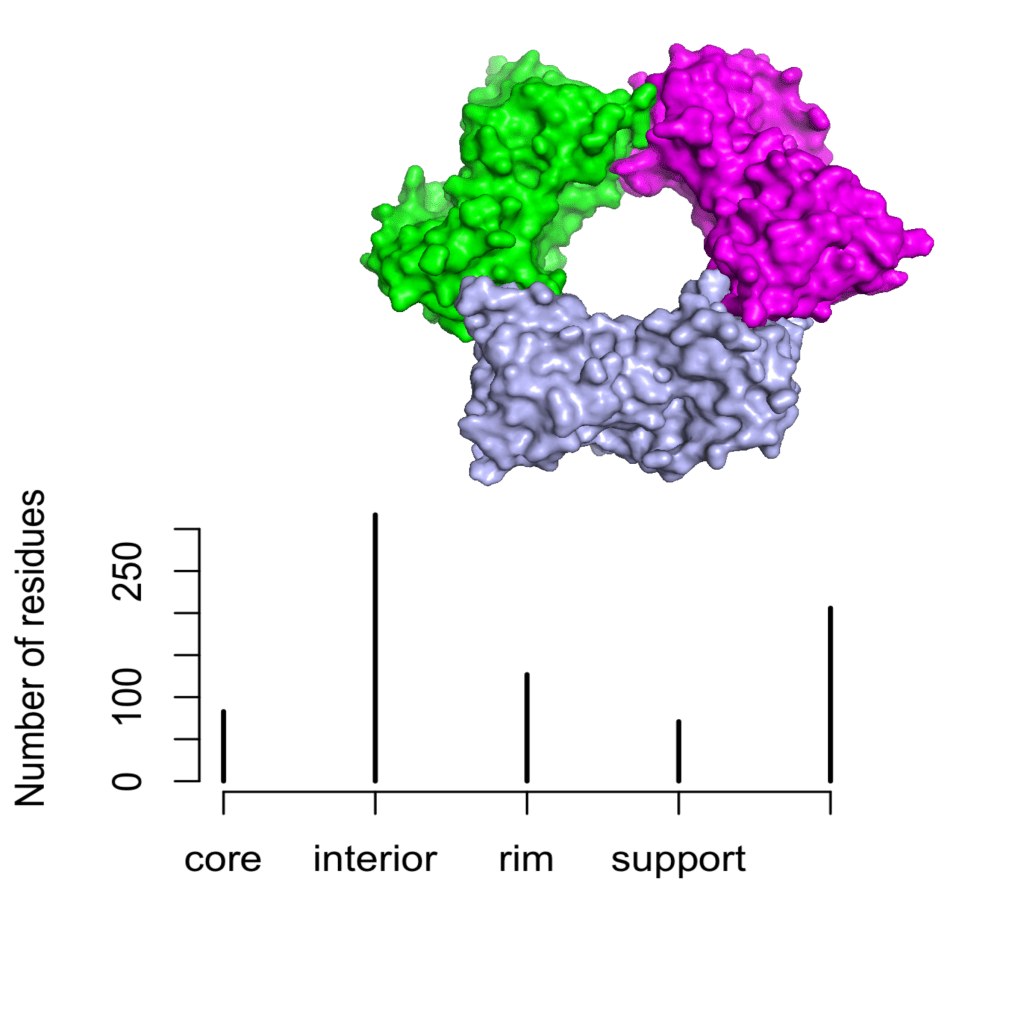
and compare the proportion of residues in each category with that of the human glyceraldehyde-3-phosphate dehydrogenase, a tetrameric protein:
# GAPDH <- stru.part(pdb = '1u8f')
# tiff(filename = "GAPDH_str", width = 10, height = 8, units = 'cm', res = 300)
# plot(table(GAPDH$str), ylab = 'Number of residues')
# dev.off()
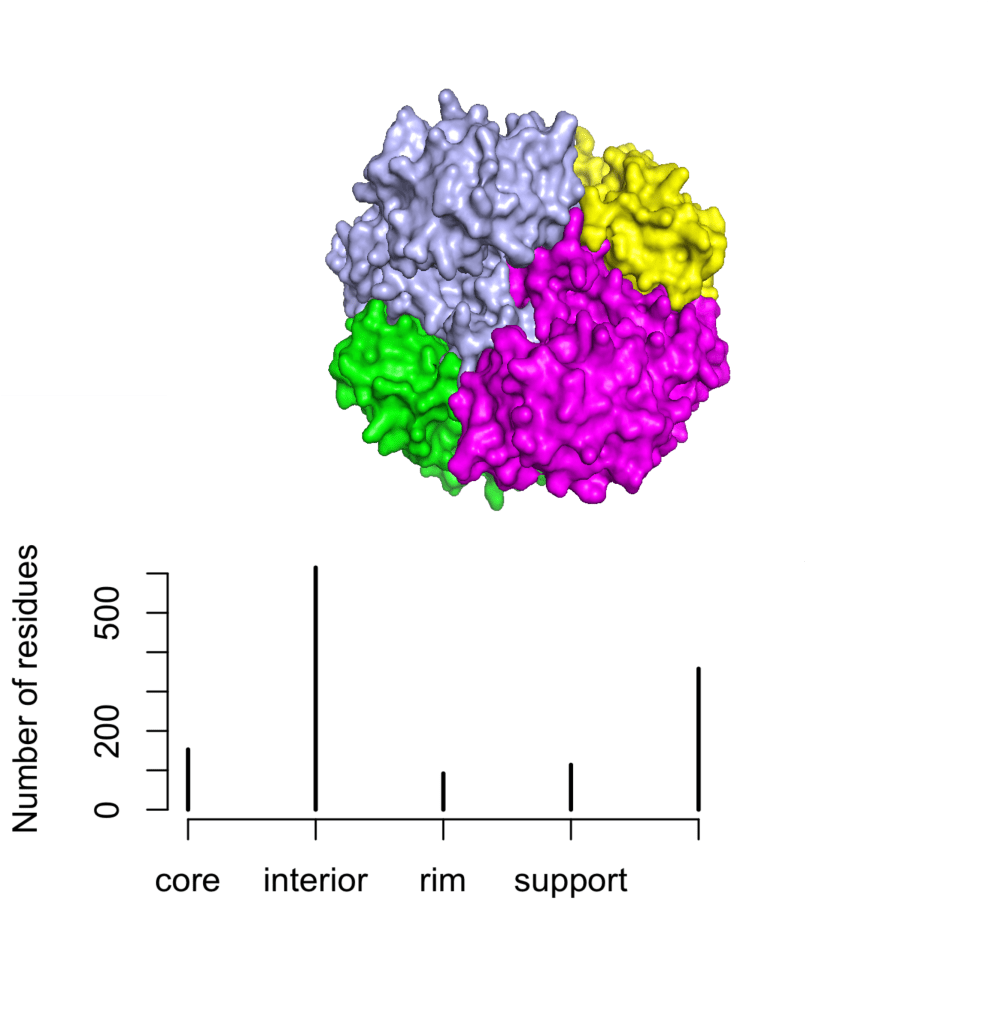
Finally, another tetrameric protein is the S-methionine adenosyltransferase from Ureaplasma urealiticum:
# uMAT <- stru.part(pdb = '6rjs')
# tiff(filename = "uMAT_str", width = 10, height = 8, units = 'cm', res = 300)
# plot(table(uMAT$str), ylab = 'Number residues')
# dev.off()